Every Saturday, my favorite radio show is "Wait Wait... Don't Tell Me! | WUNC". In yesterday's episode, the host made fun of the brand name ('Comirnaty') of Pfizer/BioNTech Covid (Listen: 46.17 minutes).
Covid vaccines from Pfizer/BioNTech, Moderna, and J&J have become the household name in just about a year or so and have been administered to millions and millions of people under FDA's Emergency Use Authorization (EUA). Under the EUA, these vaccines are just called and differentiated by the company's name: Pfizer vaccine, Moderna vaccine, and J&J vaccine. However, when the vaccine (or any drug) is formally approved, according to the regulation, it must have a brand name - in the case of Pfizer/BioNTech Covid vaccine, the brand name is now 'Comirnaty'.
The meaning behind the name 'Comirnaty': Comirnaty is an agglomeration of the words “Covid-19 immunity” and “mRNA,” the latter indicating the technology that makes the vaccine work. As a whole, the word is intended to evoke “community,”
This brings a question about the drug names: the difference between the brand name and the generic name and how to record the drug names in clinical trials. According to the CDASH, the case report form will include a form for concomitant medication including the questions like
"What was the term for the medication/therapy taken?
or
"What was the term for the medication taken?"
Concomitant medications used by the study participants prior to the trial or during the trial period should all be recorded. Usually, there is no restriction on the medication term to be recorded. Either brand name or generic name can be recorded.
The brand name of a medication is the name given by the company that makes the drug and is usually easy to say for sales and marketing purposes. The brand name may also be called the trade name. In FDA's guidance, the brand name is called proprietary name. The proprietary name of a drug product is its brand name.
The generic name, on the other hand, is the name of the active ingredient. Generic drugs are copies of brand-name drugs that have exactly the same dosage, intended use, effects, side effects, route of administration, risks, safety, and strength as the original drug. In other words, their pharmacological effects are exactly the same as those of their brand-name counterparts. In FDA's guidance, the generic name may be called 'nonproprietary name' or 'proper name' for biological products. For biological products, the term proper name means the nonproprietary name designated by FDA in the license for a biological product licensed under the PHS Act. See also 105 21 CFR 600.3(k).
For drugs that make it all the way through development, testing, and regulatory acceptance, the pharmaceutical company then gives the drug a trade name, which is a standard term in the pharmaceutical industry for a brand name, trademark name, or proprietary name. The proprietary name must be approved by the FDA and included in the product label. FDA has its guidance for industry to govern the brand name approval process "Best Practices in DevelopingProprietary Names for HumanPrescription Drug Products".
During the development stage,
sponsors of the clinical trials may use their code for the compound or drug in
the development. These drug codes will not be used when a drug is approved. For example, the famous drug from Merck 'pembrolizumab' has its own code MK-3475 that can be used in the clinical development stage and registered in clinicaltrials.gov, but will not be used as the name once the product is approved or marketed. Sponsor's drug code: MK-3475 - > generic name: pembrolizumab -> brand name: Keytruda. Similarly, Gilead's COVID-19 treatment drug has sponsor's drug code GS-5734 -> generic name: remdesivir -> Veklery. Pfizer/BioNTech's COVID-19 vaccine has sponsor's drug code: BNT16b2 -> generic name: COVID-19 Vaccine, mRNA -> brand name (proprietary name): COMIRNATY.
Here are some examples of the sponsor's drug code, generic drug name, brand drug name, and the corresponding indications.
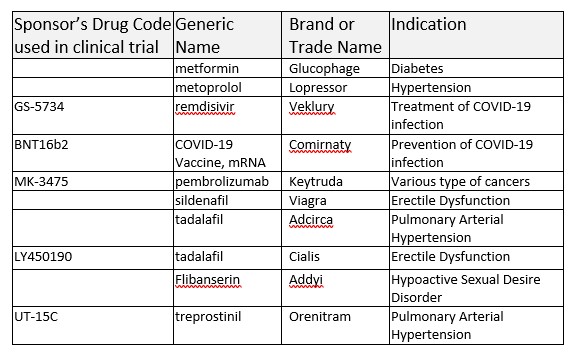
Before the statistical analyses of the concomitant medication data, the data management group will perform appropriate medical coding activities to map the recorded brand name or generic name to the corresponding ATC classifications according to WHODrug-Global dictionary.





