In a recent FDA advisory committee meeting in
November 16, 2017, we learned the first hand application of the various
approaches for multiplicity adjustment: Single step Bonferroni method, Single
step arbitrary partition of alpha level, gatekeeping - hierarchical test
procedure which was discussed in one of my previous posts.
During this meeting of
the Antimicrobial Drugs Advisory Committee (AMDAC), the committee considered
new drug application (NDA) 209367 for ciprofloxacin dry powder for inhalation
(DPI), sponsored by Bayer HealthCare Pharmaceuticals, Inc. The drug is being
proposed for the reduction of exacerbations in non-cystic fibrosis
bronchiectasis (NCFB) adult patients (≥18 years of age) with respiratory
bacterial pathogens.
The clinical program to
evaluate the safety and efficacy of ciprofloxacin DPI consisted of 2 nearly
identical phase 3, randomized, multicenter, placebo-controlled trials known as
RESPIRE 1 and RESPIRE 2. See table 1 below for the design information.
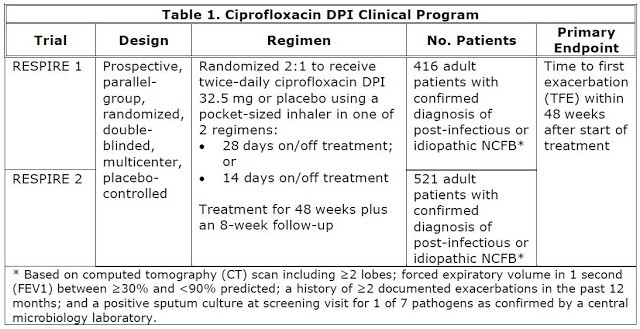
For both RESPIRE 1 and
RESPIRE 2 studies, the primary efficacy endpoint is time to first exacerbation.
Within each study, there are three treatment arms with two hypothesis tests. In
order to maintain the blinding, the placebo arm is further divided into placebo
for 28 days on/off treatment regimen and 14 days on/off treatment regimen.
However, for analysis purpose, the placebo groups are pooled. The list of
hypothesis testing and the allocated alpha are listed below. For RESPIRE 1
study, the alpha level of 0.025 for each hypothesis test is based on Bonferroni
method for multiplicity adjustment. For RESPIRE 2 study, the alpha level of
0.001 and 0.049 is based on the arbitrary partition (as long as the total alpha
= 0.05).
RESPIRE 1 Study (Bonferroni method for multiplicity adjustment):
Hypothesis
1: ciprofloxacin DPI for 28 days on/off treatment regimen versus pooled placebo
(alpha=0.025)
Hypothesis
2: ciprofloxacin DPI for 14 days on/off treatment regimen versus pooled placebo
(alpha=0.025)
RESPIRE 2 Study
(arbitrary partition of alpha level for multiplicity adjustment):
Hypothesis
1: ciprofloxacin DPI for 28 days on/off treatment regimen versus pooled placebo
(alpha=0.001)
Hypothesis
2: ciprofloxacin DPI for 14 days on/off treatment regimen versus pooled placebo
(alpha=0.049)
The study results
indicate some efficacy, but not consistent across all four hypothesis tests.
For details about the study results, please see FDA's advisory committee briefing book, study results for RESPIRE 1, and study results for RESPIRE 2 on
clinicaltrials.gov.
The study also included
a long list of the secondary efficacy endpoints. To control the overall type I
error rate associated with testing primary and secondary endpoints in two
treatment regimens (Cipro 14 and Cipro 28) against placebo, separate
hierarchical testing sequences of primary, key secondary and other secondary
endpoints were pre-specified for each regimen with statistical testing at
α=0.025 for each Cipro arm in RESPIRE 1 and α=0.001 for Cipro 28 and α=0.049
for Cipro 14 in RESPIRE 2. If the primary endpoint was significant for a Cipro
regimen then the next endpoint in the sequence (i.e., key secondary endpoint)
was tested within that Cipro regimen. Statistical testing would only continue
to the next endpoint in the hierarchy if the preceding endpoint in the
hierarchy showed significance. Endpoints which could not be statistically
tested were considered to be exploratory. The hierarchical testing strategy is
shown in Figure 2.

Unfortunately, the
hierarchical strategy did not work well and majority of the secondary endpoints
were not tested because the insignificant results in primary efficacy
endpoints. As mentioned in FDA's briefing book:
Under the pre-specified hierarchical strategy,
confirmatory testing of the first secondary endpoint (frequency of
exacerbations) against Pooled Placebo, and all subsequent endpoints, could not
be performed for Cipro 28 (both trials) and for Cipro 14 (RESPIRE 2) because
the respective findings for the primary endpoint of TFE were not significant.
In RESPIRE 1, confirmatory testing of Cipro 14 could only be performed up to
the first secondary endpoint (FOE) which failed to show significance. With the
exception of a statistically significant finding observed for one comparison
(i.e., Cipro 14 day vs. Pooled Placebo for the primary endpoint in RESPIRE 1),
all other comparisons were considered to be exploratory or not statistically
significant. As indicated in Figure 2 there was the potential for up to 32
comparisons to show statistical significance (8 endpoints in each of two Cipro
arms across two trials).
FDA advisory committee
was not convinced by the evidence of the ciprofloxacin DPI efficacy. Here is the voting result. It is
unlikely for FDA to approve a product with such a voting result even though
there is currently no approved drug for treating non-cystic fibrosis
bronchiectasis.

Had a different study
design and different method for multiplicity adjustment been used, the
situation might be very different. The evidence for the experimental drug might
be more obvious if a simpler study design was used - at least this is the
situation for 14 day on/off regimen versus placebo.
We are now closely
watching the fate of Aradigm's NDA for Ciprofloxacin in treating
non-CF bronchiectasis. Aradigm's pivital studies (Orbit
3 and Orbit
4) are simpler in study design with one of two studies positive. One
thing is for sure: there will not be the complicated situations in dealing with
the multiplicity adjustment.
References:

