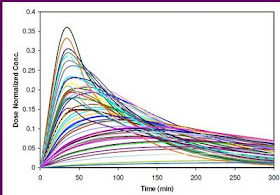
The first time when I used the term "Spaghetti" was for one of the pharmacokinetic studies where I would like to see the time-concentration curves for all individuals plotted on the same panel. The figure on the right side is an example of a Spaghetti plot from simulated data.
I don't think there is any formal definition for Spaghetti Plot, but this term refers to the plot for visualizing the trajectories for all individual subjects. The name “spaghetti plot” is called because it looks a bit like spaghetti noodles thrown on a wall.
The funny thing is that one time when I used the term 'spaghetti plot', I was asked not to use this term since it sounded like 'not formal'. Instead of using 'spaghetti plot', I had to change it to 'indivudual plots' or something like that. As a matter of fact, this term is actually used pretty often in pharmacokinetic studies and also in longitudinal studies.
In longitudinal studies, the spaghetti plot is used to visualize the trajectories or patterns or time trends. The spaghetti plot is typically used in the situation that the # of subjects is not too large and is generated for each group (if there is two treatment groups, there will be one spaghetti plot for each treatment group).
Spaghetti plot can be easily generated by software such as R and SAS. In SAS, the following statement can be used:
symbol1 value = circle color = black interpol = join repeat = 5;
proc gplot;
plot y*time = id / nolegend;
run;
Where y is the desired variable we would like to visualize; time is the time or visit; id is the subject #.
Some further readings:
1. UCLA: How can I visualize longitudinal data in SAS?
2. A oral contraceptive drug interaction study
3. A lecture notes by derived variable summaries
4. Quantitative Methods for Tracking Cognitive Change 3 Years After CABG



{kind=link}